Болезнь кори или форбса

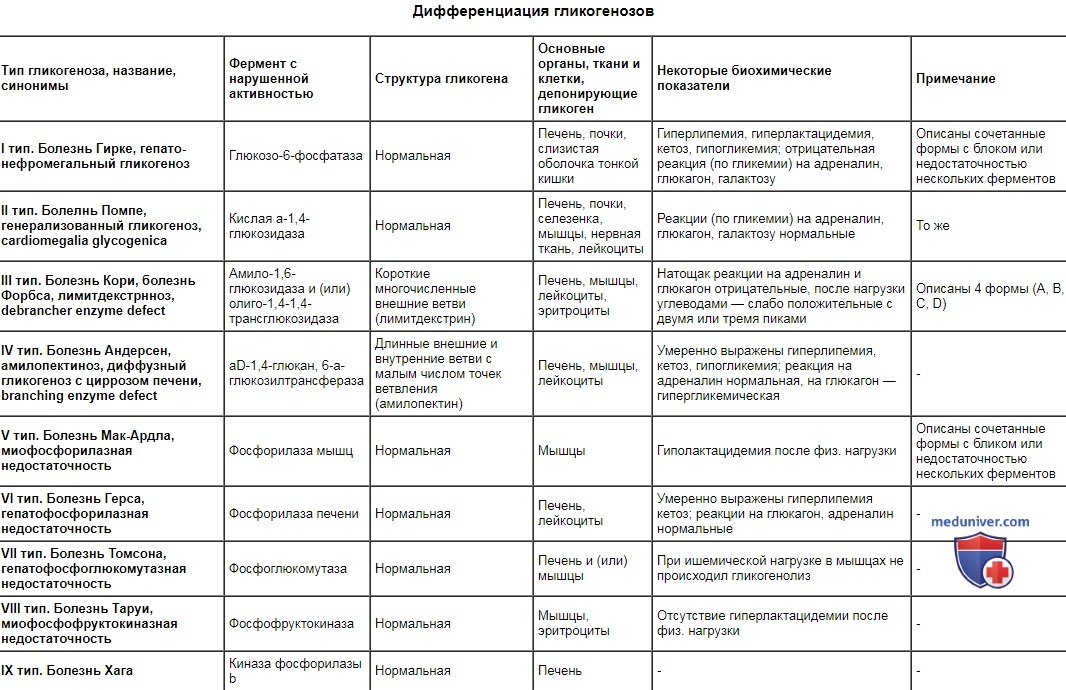
Гликогеноз I типа (болезнь Гирке) — не истинная миопатия, а заболевание, обусловленное дефицитом печеночного фермента глюкозо-6-фосфатазы, который в норме не присутствует в мышце, тем не менее у детей с этим заболеванием выявляется гипотония и умеренно выраженная мышечная слабость неизвестной этиологии. Гликогеноз II типа (болезнь Помпе) — аутосомно-рецессивно унаследованный дефицит гликолитического лизосомного фермента кислой мальтазы. Из 12 известных типов гликогенозов только II тип обусловлен дефектом лизосомного фермента. Аномальный ген картирован в локусе 17q23. Описано две формы заболевания. Младенческая форма характеризуется тяжелой генерализованной миопатией и кардиомиопатией. У пациентов выявляется кардиомегалия, гепатомегалия, диффузная гипотония и мышечная слабость. Активность КФК в крови значительно повышена. Мышечная биопсия выявляет вакуолярную миопатию в сочетании с нарушением активности ферментов лизосом, таких как кислая и щелочная фосфатазы. Смерть обычно наступает в младенческом или раннем детском возрасте. Поздняя детская или взрослая форма представлена миопатией с более легким течением, без увеличения сердца и печени. Клинические проявления могут отсутствовать до позднего детского или раннего зрелого возраста, однако возможно появление признаков мышечной слабости (обусловленной миопатией) и гипотонии даже в раннем младенческом возрасте. Активность КФК в крови значительно повышена, результаты мышечной биопсии имеют диагностическое значение даже на пресимптомной стадии заболевания. Диагноз гликогеноза II типа подтверждается при количественном анализе активности кислой мальтазы при биопсии мышц или печени. При редком варианте дефицита кислой мальтазы с легким течением ее активность при биопсии мышц может находиться на нижней границе нормы с периодическим снижением до субнормального уровня, при этом результаты мышечной биопсии напоминают гликогеноз II типа, но изменения выражены более умеренно. Другая форма — болезнь Данона — характеризуется Х-сцепленным рецессивным типом наследования, аномальный ген картирован в локусе Xql4. В основе заболевания лежит первичный дефицит протеина-2 мембран лизосом (LAMP2), который приводит к развитию гипертрофической кардиомиопатии, миопатии с поражением мышц проксимальных отделов конечностей и умственной отсталости. Гликогеноз III типа (болезнь Форбса-Кори) обусловлен дефицитом фермента, расщепляющего гликоген (амило-1,6-глюкозидаза). Это наиболее распространенный гликогеноз с наименее тяжелыми клиническими проявлениями по сравнению с другими типами гликогенозов. В младенческом возрасте часто встречаются такие симптомы, как гипотония, мышечная слабость, гепатомегалия, гипогликемия при исследовании крови натощак, однако эти симптомы часто спонтанно исчезают и в детском возрасте, а также у взрослых клинические проявления могут отсутствовать. В других случаях отмечается медленное прогрессирование атрофии мышц дистальных отделов конечностей, цирроза печени и сердечной недостаточности. При мышечной биопсии обнаруживаются минимально выраженные миопатические изменения, включающие вакуолизацию мышечных волокон. Гликогеноз IV типа (болезнь Андерсена) обусловлен дефицитом фермента, участвующего в синтезе гликогена, приводящего к синтезу аномальных молекул гликогена — амилопектина — в печени, ретикулоэндотелиальных клетках, скелетной мускулатуре и сердечной мышце. Гипотония, генерализованная мышечная слабость, атрофия мышц и контрактуры — характерные признаки миопатического процесса. Большинство пациентов умирает до 4-летнего возраста в связи с развитием печеночной или сердечной недостаточности. Описаны отдельные случаи заболевания у детей без признаков нервно-мышечного заболевания. Гликогеноз V типа (болезнь Мак-Ардла) обусловлен дефицитом мышечной фосфорилазы, наследуемым по аутосомно-рецессивному типу, аномальный ген картирован в локусе 1lql3. Основным клиническим проявлением заболевания служит непереносимость физической нагрузки, которая вызывает болезненный мышечный спазм (крампи), мышечную слабость и миоглобинурию; однако между приступами мышечная сила не снижена. Активность КФК в крови повышена только во время физической нагрузки. Характерным клиническим признаком служит отсутствие наблюдаемого в норме повышения уровня лактата в крови во время физической нагрузки, приводящей к ишемии. Это обусловлено невозможностью превращения пирувата в лактат при анаэробных состояниях in vivo. Дефицит миофосфорилазы можно обнаружить с помощью гистохимических и биохимических методов в мышечном биоптате. Редкая неонаталъная форма дефицита миофосфорилазы вызывает бульбарные расстройства в раннем младенческом возрасте, которые могут быть настолько выражены, что приводят к летальному исходу в периоде новорожденное™. В других случаях возможно медленное прогрессирование мышечной слабости, напоминающее мышечную дистрофию. Отдаленный прогноз благоприятный. Пациенты должны научиться контролировать свой уровень физической нагрузки; тяжелой инвалидизации вследствие хронической миопатии или поражения сердца не отмечается. Гликогеноз VII (болезнь Таруи) представляет собой дефицит мышечной фосфофруктокиназы. Хотя это заболевание встречается реже, чем гликогеноз V типа, оба заболевания характеризуются непереносимостью физической нагрузки, похожим клиническим течением и невозможностью превращения пирувата в лактат. Биохимическое исследование мышечных биоптатов позволяет дифференцировать эти два типа гликогенозов. Заболевание наследуется по аутосомно-рецессивному типу, аномальный ген картирован в локусе lcenq32. Дифференциация гликогенозов
 – Также рекомендуем “Митохондриальные миопатии у детей. Причины и диагностика” Оглавление темы “Нервно-мышечные болезни у детей”:
|
Источник

Гликогенозы – это группа достаточно редких наследственных заболеваний, связанных с дефектами различных ферментов, необходимых для синтеза и распада гликогена. При этом происходит накопление нормального или «неправильного» гликогена в органах и тканях человека, что и вызывает клинические проявления заболевания. Преимущественное накопление гликогена может происходить в печени, мышцах, почках. Всего описано 12 форм гликогенозов, отличие которых состоит в характере ферментной недостаточности. Прогноз у каждого вида гликогеноза свой: некоторые имеют благоприятное течение, и больные доживают до старости, другие – заканчиваются летально еще в детском возрасте. Заболевания относят к категории неизлечимых, специфическая терапия на данный момент отсутствует. Основная роль в лечении отводится диетотерапии с высоким содержанием углеводов. В этой статье мы поговорим обо всех известных медицине разновидностях гликогенозов, их симптомах и возможностях лечения.
Что такое гликоген и для чего он нужен?

Гликоген является сложным углеводом, который синтезируется путем соединения между собой молекул глюкозы, которая поступает с пищей. Он представляет собой стратегический запас глюкозы в клетках. Хранится преимущественно в печени и мышцах с той особенностью, что гликоген из печени при своем расщеплении обеспечивает глюкозой весь организм человека, а гликоген из мышц – только лишь сами мышцы. Гликоген в печени может составлять 8% от ее веса, а в мышцах – всего 1% . Но при этом за счет того, что общая мышечная масса в организме значительно больше, чем масса печени, мышечный запас превышает печеночный. Небольшое количество гликогена содержится в почках.
Как только человек приступает к какому-то роду деятельности (физическому или умственному), ему требуется энергия, которую он черпает при расщеплении гликогена и глюкозы. Поначалу расщепляется глюкоза, содержащаяся в крови, однако когда ее запасы исчерпываются (а поступления извне нет), в расход идет гликоген. Израсходованный запас гликогена затем вновь пополняется (при поступлении пищи).
Таким образом, гликоген позволяет человеку вести активную деятельность при относительно больших перерывах в еде, а не быть «привязанным к тарелке».
Этапы превращения глюкозы в гликоген и его расщепление в обратном направлении осуществляются с помощью различных ферментов, причем в печени и мышцах они различные. Нарушения деятельности таких ферментов и приводят к развитию гликогенозов.
Гликогенозы встречаются, в среднем, с частотой 1 случай на 40-68 000 населения. Они всегда носят наследственный характер, то есть возникают тогда, когда в результате генных нарушений изменяется количество или активность одного из ферментов, необходимых для биохимических процессов создания и расщепления гликогена. Тип наследования, в основном, аутосомно-рецессивный (не связан с полом, и для его появления необходимо совпадение патологических генов, полученных от отца и от матери). Из всех 12 разновидностей гликогенозов, известных на сегодняшний день, 9 являются печеночными формами, 2 – мышечными, 1 – либо мышечной, либо генерализованной (с поражением практически всего организма). У каждой из разновидностей гликогенозов имеются свои отличительные особенности.
Виды гликогенозов
Гликогеноз 0 типа (агликогеноз)
 При гликогенозе часто развиваются гипогликемические состояния, требующие введения глюкозы.
При гликогенозе часто развиваются гипогликемические состояния, требующие введения глюкозы.
Этот вид гликогеноза возникает при дефекте фермента, задействованного в создании гликогена из глюкозы, в результате чего гликоген просто не образуется в достаточном количестве. То есть возникает дефицит гликогена, поэтому этот гликогеноз стоит под нулевым номером, как бы обособленно от остальных.
При агликогенозе как только весь сахар, имеющийся в крови, израсходуется, развивается гипогликемический синдром с утратой сознания вплоть до комы. Заболевание проявляет себя практически с первых дней жизни, особенно при отсутствии у матери достаточного количества молока при грудном вскармливании. Большие перерывы между кормлениями, ночной промежуток становятся причинами развития коматозного состояния.
Кома развивается в результате отсутствия достаточного энергетического обеспечения мозга. Весьма велика вероятность смертельного исхода в раннем детском возрасте. Если же им удается выжить, то развитие таких детей, как умственное, так и физическое значительно отличается от сверстников в худшую сторону. Введение глюкозы внутривенно выводит таких больных из коматозного состояния, однако при этом довольно долго сохраняется гипергликемия (поскольку не синтезируется гликоген).
Гликогеноз I типа (болезнь Гирке)
 У таких детей может без видимой причины повышаться температура тела.
У таких детей может без видимой причины повышаться температура тела.
Источником этой разновидности является дефицит глюкозо-6-фосфатазы. Последствием становится избыточное аккумулирование гликогена в печени и почках. В крови наблюдается низкое содержание глюкозы (гипогликемия). Возникает своеобразный парадокс: гликогена избыток, но расщепить его нечем, поэтому возникает дефицит глюкозы. Больные требуют очень частых приемов пищи, чтобы концентрация глюкозы в крови была достаточной для обеспечения энергетических нужд.
Болезнь проявляет себя в первые годы жизни. У таких деток нет аппетита, возникают частые рвоты. Наблюдаются проблемы с дыханием из-за обменных нарушений: одышка, кашель. Гипогликемии могут приводить к развитию ком с судорогами. Часто повышается температура без инфекционных причин.
Откладывание гликогена в печени и почках приводит к увеличению этих органов с нарушением их функции. Из-за поражения печени развивается геморрагический синдром (склонность к спонтанным кровотечениям), нарушение фильтрационной функции почек приводит к накоплению мочевой кислоты. Если смертельный исход не настигает больных в раннем возрасте, то в последующем они отстают в физическом развитии, имеют непропорциональное тело (большая голова с «кукольным» выражением лица). Умственное развитие не страдает. Характерна гипотония и гипотрофия мышц. Половое созревание наступает значительно позже, чем у сверстников. У некоторых больных наблюдается снижение количества нейтрофилов в крови. Часто присоединяются вторичные бактериальные инфекции. Больных, которым удалось выжить и повзрослеть, настигают подагрическая нефропатия и аденомы печени. Поражение почек становится причиной потери белка с мочой и повышения артериального давления. Может возникать почечная недостаточность. Аденомы печени могут перерождаться в рак.
Гликогеноз II типа (болезнь Помпе)
Эта разновидность может быть представлена в виде двух форм: генерализованной (недостаток фермента наблюдается в печени, почках, мышцах) и мышечной (дефицит фермента только в мышцах).
Генерализованная форма дает о себе знать в первые полгода жизни. Связана с дефицитом α-глюкозидазы. Плохой аппетит, беспокойство, вялость, низкий мышечный тонус, задержка развития, нарушения дыхания становятся первыми симптомами. Постепенно увеличиваются в размерах сердце, печень, почки, селезенка. Со стороны дыхательной системы развиваются частые бронхиты и пневмонии. Развивается сердечная недостаточность. Поражение нервной системы проявляется параличами, нарушением глотания. Прогноз для жизни при генерализованной форме неблагоприятный.
Мышечная форма имеет более благоприятное течение. Является результатом дефицита кислой α-1,4-глюкозидазы только в мышцах. Заявляет о себе позже: приблизительно в 15-25 лет. Основным проявлением мышечной формы являются слабость и снижение тонуса мышц. Помимо мышечных проблем, возникают нарушения осанки (сколиотическая деформация грудного отдела позвоночника), явления незначительной сердечной недостаточности. Больные с этой формой заболевания доживают до старости.
Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз)
Это самый распространенный гликогеноз. Его причиной становится недостаточность амило-1,6-глюкозидазы, в результате чего синтезируется неправильный гликоген. Неправильный гликоген депонируется в печени, сердце и мышцах. Начальные признаки заболевания выявляются еще у младенцев. У таких детей частые рвоты, задержка в физическом развитии, «кукольное» лицо. Гипогликемии могут приводить к утрате сознания. Мышечный тонус снижается, наряду с этим наблюдается утолщение мышц, связанное с накоплением гликогена. По этой же причине утолщается сердечная мышца (гипертрофия миокарда), из-за чего нарушается сердечная проводимость и ритм сердца.
Иногда после периода полового созревания болезнь протекает менее агрессивно. При этом нарушения печени отходят на второй план, а доминирующей симптоматикой становится мышечная слабость и истончение мышц (преимущественно икроножных).
Гликогеноз IV типа (болезнь Андерсена, диффузный гликогеноз с циррозом печени, амилопектиноз)
Становится результатом дефицита амило-(1,4-1,6)-трансглюкозидазы. Это приводит к образованию неправильного гликогена. Эта разновидность гликогеноза может наследоваться сцепленно с полом, а не только аутосомно. С первых дней жизни начинается отложение неправильного гликогена в печени. Это быстро приводит к нарушению деятельности клеток печени, застою желчи, развитию гепатита, а затем и цирроза печени. Желтуха, повышенная кровоточивость, увеличение живота в размерах с накоплением жидкости в брюшной полости (асцит), кожный зуд, интоксикация организма, – все это следствия развившегося цирроза печени. Развиваются генерализованная мышечная гипотрофия и тяжелая кардиомиопатия. Часто присоединяются бактериальные инфекции. Смертельный исход наступает на 3-5 году жизни.
Гликогеноз V типа (болезнь Мак-Ардла, миофосфорилазная недостаточность)
Это исключительно мышечный гликогеноз, потому что в основе лежит изъян такого фермента, как мышечная фосфорилаза. В мышечной ткани происходит отложение нерасщепленного гликогена, из-за чего мышцы уплотняются и утолщаются, однако при этом становятся очень слабыми, быстро утомляющимися. Возникают болезненные мышечные спазмы при физической нагрузке, которые могут сопровождаться повышенной потливостью и бледностью кожных покровов, тахикардией. С мочой может выделяться мышечный белок. Все эти проявления возникают до подросткового периода и постепенно нарастают. Возможно формирование контрактур крупных суставов. По сравнению с другими разновидностями гликогенозов, гликогеноз V типа является доброкачественным заболеванием.
Гликогеноз VI типа (болезнь Герса, гепатофосфорилазная недостаточность)
В основе такого гликогеноза лежат проблемы с фосфорилазой печени. В результате гликоген накапливается в печени. Уже у младенцев наблюдается увеличение размеров печени, отмечается отставание ребенка в развитии, дети слабо набирают вес. Вместе с другими обменными нарушениями в крови выявляют повышенное содержание жира. Отмечается увеличенное содержание гликогена в красных кровяных тельцах (эритроцитах).
Гликогеноз VII типа (болезнь Таруи, миофосфофруктокиназная недостаточность)
Заболевание связано с дефицитом миофосфофруктокиназы мышц, из-за чего в них возникает отложение гликогена. По своим клиническим признакам гликогеноз VII типа практически не отличается от гликогеноза V типа и также имеет относительно доброкачественное течение.
Гликогеноз VIII типа (болезнь Томсона)
При этом гликогенозе не известна точная генетическая причина, а изъян фермента обнаружен в печени и головном мозге. На первое место выходят нарушения в нервной системе. Характерным является нистагм (непроизвольные дрожательные движения глазных яблок), что называют «танцующими глазами» в данном случае, дискоординация мышечных сокращений, что проявляется неточностью движений. Постепенно развиваются нарушение мышечного тонуса, парезы, судорожные подергивания. Неврологические расстройства неуклонно прогрессируют. Печень увеличивается в размерах, нарастают проявления печеночной недостаточности. У таких больных нет перспектив дожить до среднего возраста, заболевание заканчивается смертью в детстве.
Гликогеноз IX типа (болезнь Хага)
Это разновидность гликогеноза передается с половой хромосомой. Источником является дефицит фермента в печени. Накопление гликогена приводит к печеночной недостаточности.
Гликогеноз X типа
Эта разновидность описана всего лишь единственный раз во всем мире. Тип наследования установить не удалось. Заболевание протекало с увеличением печени, сопровождалось болью и напряжением мышц при вовлечении их в работу.
Гликогеноз XI типа (болезнь Фанкони-Бикеля)
Гликогеноз с неустановленным механизмом передачи. Ферментные дефекты обнаружены в печени и почках. Этой разновидности гликогенозов свойственно увеличение размеров и уплотнение печени, отставание в росте. Отличием от других разновидностей гликогенозов является уменьшение количества фосфатов в крови и развитие в связи с этим рахита. По достижении периода полового созревания наблюдается тенденция к некоторому улучшению состояния: печень уменьшается в размерах, содержание фосфора нормализуется, дети начинают расти.
Лечение
Кукурузный крахмал обеспечивает ребенка глюкозой на несколько часов, это помогает избежать гипогликемии в ночное время.
Гликогенозы, как и практически все генетические заболевания, являются неизлечимой патологией. Все меры медицинской помощи, по существу, являются симптоматическими. Тем не менее, поскольку ряд гликогенозов имеет благоприятный прогноз для жизни при соблюдении ряда условий (в частности мышечная форма II типа, III, V, VI, VII, IX, XI тип), то лечебные мероприятия способствуют уменьшению ряда симптомов и улучшению состояния здоровья пациента.
В основу лечения при гликогенозах положена диетотерапия, позволяющая избежать гипогликемии и второстепенных нарушений метаболических процессов в организме. Суть диеты заключается в изучении гликемического профиля больного и подборе такого режима приема пищи, который позволит избежать прогрессирования биохимических нарушений (нарушений метаболизма жиров, молочной кислоты) и обеспечит достаточный уровень глюкозы в крови. Частые, в том числе ночные, кормления у маленьких детей помогают избежать гипогликемии. Обычно назначается пища, содержащая много белков и углеводов, а жиры ограничиваются. Процентное соотношение приблизительно следующее: углеводы — 70%, белки – 10%, жиры – 20%.
Для того чтобы не приходилось кормить ребенка несколько раз за ночь, может использоваться сырой кукурузный крахмал (назначается детям старше 1 года), который разводят водой в соотношении 1:2. Начинают введение с дозы 0,25 мг/кг, затем ее постепенно увеличивают настолько, чтобы введенной дозы крахмала хватало для обеспечения организма глюкозой на 6-8 часов, то есть на всю ночь. Таким образом, прием крахмала на ночь позволяет отказаться от ночных кормлений, что обеспечивает детям полноценный сон без перерывов.
В тех случаях, когда маленькие дети страдают от частых приступов гипогликемии, и повлиять на это только соблюдением диеты не удается, назначается дополнительное введение чистой глюкозы или смеси, обогащенной мальтодекстрином.
При гликогенозе I типа требуется значительно ограничить продукты, содержащие галактозу и фруктозу (молоко, большинство фруктов). При III типе гликогеноза таких ограничений нет. При VII типе нужно ограничить поступление сахарозы.
В ряде случаев (особенно при возникновении других, интеркуррентных заболеваний у таких детей) одного энтерального питания становится недостаточно, поскольку потребность организма в энергии повышается. Тогда прибегают к кормлению через назогастральный зонд и внутривенным инфузиям в условиях стационара.
Те разновидности гликогенозов, при которых дефекты ферментов локализованы только в мышцах, требуют употребления фруктозы внутрь по 50-100 г в день, комплекса витаминов, аденозинтрифосфорной кислоты.
Из медикаментозных препаратов при гликогенозе I типа используют препараты кальция, витамин Д и В1, аллопуринол (для предотвращения подагры и отложения уратов в почках), никотиновую кислоту (для снижения риска калькулезного холецистита и предотвращения панкреатита). Если с почками начинает выводиться белок, тогда назначают ингибиторы ангиотензинпревращающего фермента (Лизиноприл, Эналаприл и другие).
Для гликогеноза II типа разработана специфическая ферментная терапия (заместительная). Препарат Миозим вводится по 20 мг/кг каждые две недели. Миозим представляет собой созданный с помощью генной инженерии искусственный человеческий фермент α-глюкозидазу. Естественно, эффект тем больше, чем раньше начато лечение. Но пока что препарат разрешен к применению только в некоторых странах Европы, в Японии и США. Генная инженерия продолжает разработки в этом направлении, пытаясь синтезировать и другие ферменты, необходимые для нормального синтеза и расщепления гликогена, чтобы помочь больным с остальными формами гликогенозов.
Некоторым больным помогает введение глюкокортикоидов, анаболических гормонов и глюкагона. Препараты стимулируют некоторые биохимические процессы (например, глюконеогенез, то есть процесс синтеза глюкозы из неуглеводных веществ), тем самым уменьшая проявления заболевания.
Из хирургических методов лечения при некоторых формах гликогенозов используют наложение портокавального анастомоза или трансплантацию печени. Портокавальный анастомоз накладывают больным с тяжелой формой гликогеноза I и III типов. Он позволяет уменьшить обменные нарушения, способствует регрессу размеров печени, улучшает переносимость гипогликемии. Пересадка печени от донора осуществляется при I, III, IV типах гликогенозов. При гликогенозе I типа операция проводится только при неэффективности мероприятий диетотерапии, при гликогенозе III типа – когда печень больного уже не спасти.
Таким образом, гликогенозы – это довольно обширная группа болезней обмена веществ с генетическими истоками. На сегодняшний день, медицина не располагает 100%- ми методами эффективного лечения сего недуга, перспективы в этом направлении принадлежат генной инженерии.
Источник