Болезнь кори и форбса

А
Б
В
Г
Д
Ж
З
И
Й
К
Л
М
Н
О
П
Р
С
Т
У
Ф
Х
Ц
Ч
Ш
Э
Ю
Я
- Что такое Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз)
- Что провоцирует Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз)
- Патогенез (что происходит?) во время Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
- Симптомы Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
- Диагностика Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
- Лечение Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
- К каким докторам следует обращаться если у Вас Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз)
Что такое Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз)
Гликогеноз третьего типа связан с мутациями структурного гена цитозольной амило-1,6-глюкозидазы, экспрессирующейся во многих тканях: печени, мышцах, эритроцитах. Ген картирован на хромосоме 1р21. Как и предыдущие варианты, третий тип гликогеноза наследуется по аутосомно-рецессивному типу.
Что провоцирует Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз)
Гликогеноз III наследуется по аутосомно-рецессивному типу. Нередко родители состоят в кровном родстве. В одной семье иногда болеют брат и сестра или два брата. В основе заболевания лежит мутация гена, которая выявляется клинически у гомозиготов.
Патогенез (что происходит?) во время Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
В популяции евреев-сефардов (выходцев из Северной Африки) болезнь встречается с частотой 1:5400 новорожденных. Амило-1,6-глкжозидаза участвует в метаболизме гликогена в точках ветвления гликогенового «дерева». Фермент является бифункциональным: с одной стороны, превращает лимит-декстрин в гликоген с наружными цепями нормальной длины и, с другой стороны, освобождает глюкозу путем гидролиза а-1,6-глюкозидной связи. Недостаточность фермента приводит к нарушению гликогенолиза и накоплению в тканях молекул гликогена аномальной формы с укороченными наружными цепями. Также как и при гликогенозах 1 и 2 типов, при этом варианте заболевания нарушение гликогенолиза сопровождается гипогликемией, лактат-ацидозом, гиперкетонемией.
Патологическая анатомия. В печени, мышцах и сердце происходит накопление гликогена. При химическом исследовании обнаруживается аномалия структуры гликогена (лимитдекстрин). Гистологически выявляются большие набухшие фибриллы, подвергшиеся вакуолизации. Гепатоциты вакуолизированы и выглядят пенистыми, а в портальных пространствах отмечаются фиброз и круглоклеточная инфильтрация.
Электронно-микроскопически гликоген печени выявляется в виде альфа- и бета-частичек, клеточные органеллы нормальны и находятся вне скоплений гликогена. Клинически этот тип гликогеноза напоминает I тип, однако симптомы не столь резко выражены. Дети невысокого роста, с кукольным лицом и большим животом.
Отмечается увеличение подкожной жировой клетчатки на лице и на туловище, в связи с чем конечности выглядят тонкими. Важным клиническим симптомом является значительная гепатомегалия, которая отмечается уже на первом-втором месяце жизни. Печень быстро увеличивается и занимает – брюшной полости.
Симптомы Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
Клинические проявления варьируют.
Различают две клинические формы с хроническим течением:
- IIIа- при которой возникают симптомы поражения печении мышц и I
- IIIb – при которой поражается только печень.
Заболевание начинается в возрасте от 6 мес до 3-х лет. Клиническая картина в детском возрасте сходна с таковой при гликогенозе I типа. Характерные симптомы – гепатомегалия, отставание в росте, гипотрофия, «кукольное» лицо, локальные отложения жира, кожные ксантомы. Типичны лактат-ацидоз с гиперкетонемией при голодании. Гепатомегалия с дисфункцией печени, найденная у всех больных в детстве, имеет тенденцию исчезать в постпубертатный период. Больные обычно доживают до взрослого возраста, но возможен синдром внезапной смерти младенца. У взрослых больных доминирует миопатия с прогрессирующей мышечной слабостью при физической нагрузке (иногда в виде шаткой походки), гипотрофия мышц дистальных отделов нижних конечностей (преимущественно икроножных мышц), в более позднем возрасте присоединяется поражение мышц рук.
Диагностика Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
Диагностика заболевания проводится на основе клинической картины и данных лабораторного исследования: снижения активности амило- 1,6-глюкозидазы и отложения гликогена измененной структуры в гепатоцитах и мышцах. В плазме крови отмечается увеличение концентрации лактата, мочевой кислоты, холестерина и триглицеридов.
Лечение Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
Из-за нарушения гликогенолиза при гликогенозе типа III продукция глюкозы недостаточна, поэтому у грудных детей и детей младшего возраста после ночного голодания возникает гипогликемия. Усиление глюконеогенеза приводит к снижению уровня аминокислот в плазме (они используются как субстраты глюконеогенеза).
Таким образом, цель лечения – предупредить гипогликемию голодания и возместить дефицит аминокислот. Проводится оно следующим образом:
- прием необходимого количества глюкозы в виде сырого кукурузного крахмала в сочетании с диетой, содержащей достаточное количество белков и других питательных веществ, устраняет метаболические нарушения и задержку роста;
- больным с выраженной задержкой роста и тяжелой миопатией показано непрерывное ночное зондовое питание смесью, содержащей глюкозу, олигосахариды и аминокислоты, и частый прием богатой белком пищи в дневное время.
К каким докторам следует обращаться если у Вас Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз)
Генетик
Педиатр
А
Б
В
Г
Д
Ж
З
И
Й
К
Л
М
Н
О
П
Р
С
Т
У
Ф
Х
Ц
Ч
Ш
Э
Ю
Я
Акции и специальные предложения
Медицинские новости
21.09.2020
Специалисты заявляют о нестабильной эпидемиологической ситуации по заболеваемости коклюшем в различных регионах РФ – в том числе, и в Санкт-Петербурге. По данным Федеральной службы по надзору…
07.07.2020

Мигрень широко распространена во всем мире, изучена лучше других типов головной боли и является второй ведущей причиной потерянных лет жизни . На сегодняшний день в России от этого заболевания страдают более 20 миллионов человек. При этом большинство из них не знают о своем диагнозе…
01.06.2020
Фотовыставка «Видеть главное», посвященная пациентам с псориазом, открылась на портале МБОО «Кожные и аллергические болезни» в виртуальном формате. «Видеть главное» — это 12 портретов, выполненных в технике стерео-варио, которая позволяет увидеть фото со следами псориаза и без них в зависимости от того, под каким углом смотрит посетитель.
23.05.2020
Статистика показывает, что ишемическая болезнь сердца и инсульт уносят больше всего человеческих жизней во всем мире. Коронавирус COVID-19 – серьезное явление, но про здоровье других органов в тоже время забывать не стоит. На здоровье сердца влияет не так много факторов…
Медицинские статьи

Офтальмология является одной из наиболее динамично развивающихся областей медицины. Ежегодно появляются технологии и процедуры, позволяющие получать результат, который еще 5–10 лет назад казался недостижимым. К примеру, в начале XXI века лечение возрастной дальнозоркости было невозможно. Максимум, на что мог рассчитывать пожилой пациент, — это на…
Почти 5% всех злокачественных опухолей составляют саркомы. Они отличаются высокой агрессивностью, быстрым распространением гематогенным путем и склонностью к рецидивам после лечения. Некоторые саркомы развиваются годами, ничем себя не проявляя…
Вирусы не только витают в воздухе, но и могут попадать на поручни, сидения и другие поверхности, при этом сохраняя свою активность. Поэтому в поездках или общественных местах желательно не только исключить общение с окружающими людьми, но и избегать…
Вернуть хорошее зрение и навсегда распрощаться с очками и контактными линзами – мечта многих людей. Сейчас её можно сделать реальностью быстро и безопасно. Новые возможности лазерной коррекции зрения открывает полностью бесконтактная методика Фемто-ЛАСИК.
Источник
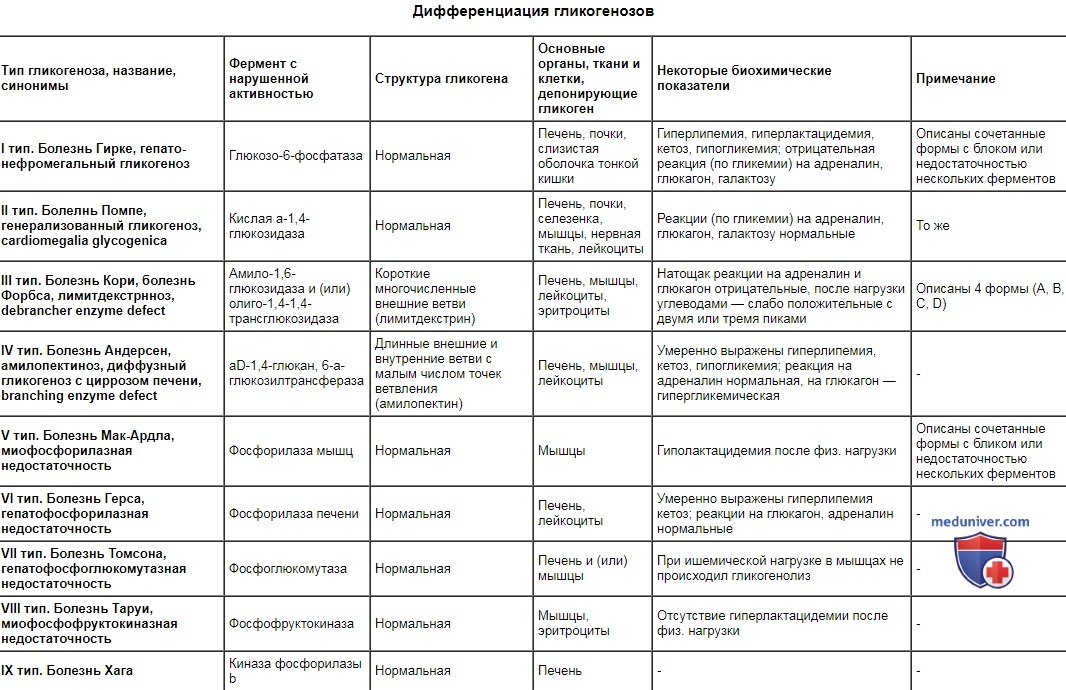
Гликогеноз I типа (болезнь Гирке) — не истинная миопатия, а заболевание, обусловленное дефицитом печеночного фермента глюкозо-6-фосфатазы, который в норме не присутствует в мышце, тем не менее у детей с этим заболеванием выявляется гипотония и умеренно выраженная мышечная слабость неизвестной этиологии. Гликогеноз II типа (болезнь Помпе) — аутосомно-рецессивно унаследованный дефицит гликолитического лизосомного фермента кислой мальтазы. Из 12 известных типов гликогенозов только II тип обусловлен дефектом лизосомного фермента. Аномальный ген картирован в локусе 17q23. Описано две формы заболевания. Младенческая форма характеризуется тяжелой генерализованной миопатией и кардиомиопатией. У пациентов выявляется кардиомегалия, гепатомегалия, диффузная гипотония и мышечная слабость. Активность КФК в крови значительно повышена. Мышечная биопсия выявляет вакуолярную миопатию в сочетании с нарушением активности ферментов лизосом, таких как кислая и щелочная фосфатазы. Смерть обычно наступает в младенческом или раннем детском возрасте. Поздняя детская или взрослая форма представлена миопатией с более легким течением, без увеличения сердца и печени. Клинические проявления могут отсутствовать до позднего детского или раннего зрелого возраста, однако возможно появление признаков мышечной слабости (обусловленной миопатией) и гипотонии даже в раннем младенческом возрасте. Активность КФК в крови значительно повышена, результаты мышечной биопсии имеют диагностическое значение даже на пресимптомной стадии заболевания. Диагноз гликогеноза II типа подтверждается при количественном анализе активности кислой мальтазы при биопсии мышц или печени. При редком варианте дефицита кислой мальтазы с легким течением ее активность при биопсии мышц может находиться на нижней границе нормы с периодическим снижением до субнормального уровня, при этом результаты мышечной биопсии напоминают гликогеноз II типа, но изменения выражены более умеренно. Другая форма — болезнь Данона — характеризуется Х-сцепленным рецессивным типом наследования, аномальный ген картирован в локусе Xql4. В основе заболевания лежит первичный дефицит протеина-2 мембран лизосом (LAMP2), который приводит к развитию гипертрофической кардиомиопатии, миопатии с поражением мышц проксимальных отделов конечностей и умственной отсталости. Гликогеноз III типа (болезнь Форбса-Кори) обусловлен дефицитом фермента, расщепляющего гликоген (амило-1,6-глюкозидаза). Это наиболее распространенный гликогеноз с наименее тяжелыми клиническими проявлениями по сравнению с другими типами гликогенозов. В младенческом возрасте часто встречаются такие симптомы, как гипотония, мышечная слабость, гепатомегалия, гипогликемия при исследовании крови натощак, однако эти симптомы часто спонтанно исчезают и в детском возрасте, а также у взрослых клинические проявления могут отсутствовать. В других случаях отмечается медленное прогрессирование атрофии мышц дистальных отделов конечностей, цирроза печени и сердечной недостаточности. При мышечной биопсии обнаруживаются минимально выраженные миопатические изменения, включающие вакуолизацию мышечных волокон. Гликогеноз IV типа (болезнь Андерсена) обусловлен дефицитом фермента, участвующего в синтезе гликогена, приводящего к синтезу аномальных молекул гликогена — амилопектина — в печени, ретикулоэндотелиальных клетках, скелетной мускулатуре и сердечной мышце. Гипотония, генерализованная мышечная слабость, атрофия мышц и контрактуры — характерные признаки миопатического процесса. Большинство пациентов умирает до 4-летнего возраста в связи с развитием печеночной или сердечной недостаточности. Описаны отдельные случаи заболевания у детей без признаков нервно-мышечного заболевания. Гликогеноз V типа (болезнь Мак-Ардла) обусловлен дефицитом мышечной фосфорилазы, наследуемым по аутосомно-рецессивному типу, аномальный ген картирован в локусе 1lql3. Основным клиническим проявлением заболевания служит непереносимость физической нагрузки, которая вызывает болезненный мышечный спазм (крампи), мышечную слабость и миоглобинурию; однако между приступами мышечная сила не снижена. Активность КФК в крови повышена только во время физической нагрузки. Характерным клиническим признаком служит отсутствие наблюдаемого в норме повышения уровня лактата в крови во время физической нагрузки, приводящей к ишемии. Это обусловлено невозможностью превращения пирувата в лактат при анаэробных состояниях in vivo. Дефицит миофосфорилазы можно обнаружить с помощью гистохимических и биохимических методов в мышечном биоптате. Редкая неонаталъная форма дефицита миофосфорилазы вызывает бульбарные расстройства в раннем младенческом возрасте, которые могут быть настолько выражены, что приводят к летальному исходу в периоде новорожденное™. В других случаях возможно медленное прогрессирование мышечной слабости, напоминающее мышечную дистрофию. Отдаленный прогноз благоприятный. Пациенты должны научиться контролировать свой уровень физической нагрузки; тяжелой инвалидизации вследствие хронической миопатии или поражения сердца не отмечается. Гликогеноз VII (болезнь Таруи) представляет собой дефицит мышечной фосфофруктокиназы. Хотя это заболевание встречается реже, чем гликогеноз V типа, оба заболевания характеризуются непереносимостью физической нагрузки, похожим клиническим течением и невозможностью превращения пирувата в лактат. Биохимическое исследование мышечных биоптатов позволяет дифференцировать эти два типа гликогенозов. Заболевание наследуется по аутосомно-рецессивному типу, аномальный ген картирован в локусе lcenq32. Дифференциация гликогенозов
 – Также рекомендуем “Митохондриальные миопатии у детей. Причины и диагностика” Оглавление темы “Нервно-мышечные болезни у детей”:
|
Источник
- Что такое Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз)
- Что провоцирует / Причины Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
- Патогенез (что происходит?) во время Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
- Симптомы Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
- Диагностика Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
- Лечение Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза)
- К каким докторам следует обращаться если у Вас Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз)
Что такое Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз) –
Гликогеноз третьего типа связан с мутациями структурного гена цитозольной амило-1,6-глюкозидазы, экспрессирующейся во многих тканях: печени, мышцах, эритроцитах. Ген картирован на хромосоме 1р21. Как и предыдущие варианты, третий тип гликогеноза наследуется по аутосомно-рецессивному типу.
Что провоцирует / Причины Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза):
Гликогеноз III наследуется по аутосомно-рецессивному типу. Нередко родители состоят в кровном родстве. В одной семье иногда болеют брат и сестра или два брата. В основе заболевания лежит мутация гена, которая выявляется клинически у гомозиготов.
Патогенез (что происходит?) во время Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза):
В популяции евреев-сефардов (выходцев из Северной Африки) болезнь встречается с частотой 1:5400 новорожденных. Амило-1,6-глкжозидаза участвует в метаболизме гликогена в точках ветвления гликогенового «дерева». Фермент является бифункциональным: с одной стороны, превращает лимит-декстрин в гликоген с наружными цепями нормальной длины и, с другой стороны, освобождает глюкозу путем гидролиза а-1,6-глюкозидной связи. Недостаточность фермента приводит к нарушению гликогенолиза и накоплению в тканях молекул гликогена аномальной формы с укороченными наружными цепями. Также как и при гликогенозах 1 и 2 типов, при этом варианте заболевания нарушение гликогенолиза сопровождается гипогликемией, лактат-ацидозом, гиперкетонемией.
Патологическая анатомия. В печени, мышцах и сердце происходит накопление гликогена. При химическом исследовании обнаруживается аномалия структуры гликогена (лимитдекстрин). Гистологически выявляются большие набухшие фибриллы, подвергшиеся вакуолизации. Гепатоциты вакуолизированы и выглядят пенистыми, а в портальных пространствах отмечаются фиброз и круглоклеточная инфильтрация.
Электронно-микроскопически гликоген печени выявляется в виде альфа- и бета-частичек, клеточные органеллы нормальны и находятся вне скоплений гликогена. Клинически этот тип гликогеноза напоминает I тип, однако симптомы не столь резко выражены. Дети невысокого роста, с кукольным лицом и большим животом.
Отмечается увеличение подкожной жировой клетчатки на лице и на туловище, в связи с чем конечности выглядят тонкими. Важным клиническим симптомом является значительная гепатомегалия, которая отмечается уже на первом-втором месяце жизни. Печень быстро увеличивается и занимает – брюшной полости.
Симптомы Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза):
Клинические проявления варьируют.
Различают две клинические формы с хроническим течением:
- IIIа- при которой возникают симптомы поражения печении мышц и I
- IIIb – при которой поражается только печень.
Заболевание начинается в возрасте от 6 мес до 3-х лет. Клиническая картина в детском возрасте сходна с таковой при гликогенозе I типа. Характерные симптомы – гепатомегалия, отставание в росте, гипотрофия, «кукольное» лицо, локальные отложения жира, кожные ксантомы. Типичны лактат-ацидоз с гиперкетонемией при голодании. Гепатомегалия с дисфункцией печени, найденная у всех больных в детстве, имеет тенденцию исчезать в постпубертатный период. Больные обычно доживают до взрослого возраста, но возможен синдром внезапной смерти младенца. У взрослых больных доминирует миопатия с прогрессирующей мышечной слабостью при физической нагрузке (иногда в виде шаткой походки), гипотрофия мышц дистальных отделов нижних конечностей (преимущественно икроножных мышц), в более позднем возрасте присоединяется поражение мышц рук.
Диагностика Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза):
Диагностика заболевания проводится на основе клинической картины и данных лабораторного исследования: снижения активности амило- 1,6-глюкозидазы и отложения гликогена измененной структуры в гепатоцитах и мышцах. В плазме крови отмечается увеличение концентрации лактата, мочевой кислоты, холестерина и триглицеридов.
Лечение Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза):
Из-за нарушения гликогенолиза при гликогенозе типа III продукция глюкозы недостаточна, поэтому у грудных детей и детей младшего возраста после ночного голодания возникает гипогликемия. Усиление глюконеогенеза приводит к снижению уровня аминокислот в плазме (они используются как субстраты глюконеогенеза).
Таким образом, цель лечения – предупредить гипогликемию голодания и возместить дефицит аминокислот. Проводится оно следующим образом:
- прием необходимого количества глюкозы в виде сырого кукурузного крахмала в сочетании с диетой, содержащей достаточное количество белков и других питательных веществ, устраняет метаболические нарушения и задержку роста;
- больным с выраженной задержкой роста и тяжелой миопатией показано непрерывное ночное зондовое питание смесью, содержащей глюкозу, олигосахариды и аминокислоты, и частый прием богатой белком пищи в дневное время.
К каким докторам следует обращаться если у Вас Гликогеноз III типа (болезнь Кори, болезнь Форбса, лимитдекстриноз):
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Гликогеноза III типа (болезни Кори, болезни Форбса, лимитдекстриноза), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу.
Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
У Вас ? Необходимо очень тщательно подходить к состоянию Вашего здоровья в целом. Люди уделяют недостаточно внимания симптомам заболеваний и не осознают, что эти болезни могут быть жизненно опасными. Есть много болезней, которые по началу никак не проявляют себя в нашем организме, но в итоге оказывается, что, к сожалению, их уже лечить слишком поздно. Каждое заболевание имеет свои определенные признаки, характерные внешние проявления – так называемые симптомы болезни. Определение симптомов – первый шаг в диагностике заболеваний в целом. Для этого просто необходимо по несколько раз в год проходить обследование у врача, чтобы не только предотвратить страшную болезнь, но и поддерживать здоровый дух в теле и организме в целом.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации, возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой. Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе Вся медицина. Также зарегистрируйтесь на медицинском портале Eurolab, чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
Другие заболевания из группы Болезни эндокринной системы, расстройства питания и нарушения обмена веществ:
| Аддисонический криз (острая недостаточность коры надпочечников) |
| Аденома молочной железы |
| Адипозогенитальная дистрофия (болезнь Перхкранца – Бабинского – Фрелиха) |
| Адреногенитальный синдром |
| Акромегалия |
| Алиментарный маразм (алиментарная дистрофия) |
| Алкалоз |
| Алкаптонурия |
| Амилоидоз (амилоидная дистрофия) |
| Амилоидоз желудка |
| Амилоидоз кишечника |
| Амилоидоз островков поджелудочной железы |
| Амилоидоз печени |
| Амилоидоз пищевода |
| Ацидоз |
| Белково-энергетическая недостаточность |
| Болезнь I-клеток (муколипидоз типа II) |
| Болезнь Вильсона-Коновалова (гепатоцеребральная дистрофия) |
| Болезнь Гоше (глюкоцереброзидный липидоз, глюкоцереброзидоз) |
| Болезнь Иценко-Кушинга |
| Болезнь Краббе (глобоидно-клеточная лейкодистрофия) |
| Болезнь Нимана – Пика (сфингомиелиноз) |
| Болезнь Фабри |
| Ганглиозидоз GM1 тип I |
| Ганглиозидоз GM1 тип II |
| Ганглиозидоз GM1 тип III |
| Ганглиозидоз GM2 |
| Ганглиозидоз GM2 тип I (амавротическая идиотия Тея – Сакса, болезнь Тея – Сакса) |
| Ганглиозидоз GM2 тип II (болезнь Сандхоффа, амавротическая идиотия Сандхоффа) |
| Ганглиозидоз GM2 ювенильный |
| Гигантизм |
| Гиперальдостеронизм |
| Гиперальдостеронизм вторичный |
| Гиперальдостеронизм первичный (синдром Конна) |
| Гипервитаминоз D |
| Гипервитаминоз А |
| Гипервитаминоз Е |
| Гиперволемия |
| Гипергликемическая (диабетическая) кома |
| Гиперкалиемия |
| Гиперкальциемия |
| Гиперлипопротеинемия I типа |
| Гиперлипопротеинемия II типа |
| Гиперлипопротеинемия III типа |
| Гиперлипопротеинемия IV типа |
| Гиперлипопротеинемия V типа |
| Гиперосмолярная кома |
| Гиперпаратиреоз вторичный |
| Гиперпаратиреоз первичный |
| Гиперплазия тимуса (вилочковой железы) |
| Гиперпролактинемия |
| Гиперфункция яичек |
| Гиперхолестеринемия |
| Гиповолемия |
| Гипогликемическая кома |
| Гипогонадизм |
| Гипогонадизм гиперпролактинемический |
| Гипогонадизм изолированный (идиопатический) |
| Гипогонадизм первичный врожденный (анорхизм) |
| Гипогонадизм первичный приобретенный |
| Гипокалиемия |
| Гипопаратиреоз |
| Гипопитуитаризм |
| Гипотиреоз |
| Гликогеноз 0 типа (агликогеноз) |
| Гликогеноз I типа (болезнь Гирке) |
| Гликогеноз II типа (болезнь Помпе) |
| Гликогеноз IV типа (болезнь Андерсена, амилопектиноз, диффузный гликогеноз с циррозом печени) |
| Гликогеноз IX типа (болезнь Хага) |
| Гликогеноз V типа (болезнь Мак-Ардла, миофосфорилазная недостаточность) |
| Гликогеноз VI типа (болезнь Герса, гепатофосфорилазная недостаточность) |
| Гликогеноз VII типа (болезнь Таруи, миофосфофруктокиназная недостаточность) |
| Гликогеноз VIII типа (болезнь Томсона) |
| Гликогеноз XI типа |
| Гликогеноз Х типа |
| Дефицит (недостаточность) ванадия |
| Дефицит (недостаточность) магния |
| Дефицит (недостаточность) марганца |
| Дефицит (недостаточность) меди |
| Дефицит (недостаточность) молибдена |
| Дефицит (недостаточность) хрома |
| Дефицит железа |
| Дефицит кальция (алиментарная недостаточность кальция) |
| Дефицит цинка (алиментарная недостаточность цинка) |
| Диабетическая кетоацидотическая кома |
| Дисфункция яичников |
| Диффузный (эндемический) зоб |
| Задержка полового созревания |
| Избыток эстрогенов |
| Инволюция молочных желез |
| Карликовость (низкорослость) |
| Квашиоркор |
| Кистозная мастопатия |
| Ксантинурия |
| Лактацидемическая кома |
| Лейциноз (болезнь кленового сиропа) |
| Липидозы |
| Липогранулематоз Фарбера |
| Липодистрофия (жировая дистрофия) |
| Липодистрофия врожденная генерализованная (синдром Сейпа-Лоуренса) |
| Липодистрофия гипермускулярная |
| Липодистрофия постинъекционная |
| Липодистрофия прогрессирующая сегментарная |
| Липоматоз |
| Липоматоз болезненный |
| Метахроматическая лейкодистрофия |
| Микседематозная кома |
| Муковисцидоз (кистозный фиброз) |
| Мукополисахаридоз |
| Мукополисахаридоз типа I-S (болезнь Шейе; поздний синдром Гурлер) |
| Мукополисахаридоз типа I-Н (синдром Гурлер) |
| Мукополисахаридоз типа II (синдром Гунтера) |
| Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо) |
| Мукополисахаридоз типа IV (снидром Моркио, болезнь Моркио). |
| Мукополисахаридоз типа VI (синдром Марото-Лами, болезнь Марото-Лами) |
| Мукополисахаридоз типа VII (синдром Слая) |
| Мукополисахаридоз типа VIII (синдром Ди Ферранте) |
| Надпочечниковая гиперандрогения |
| Нарушение обмена тирозина |
| Недостаточность аскорбиновой кислоты ( гиповитаминоз С) |
| Недостаточность витамина B1 (тиамина) |
| Недостаточность витамина D |
| Недостаточность витамина А |
| Недостаточность витамина В12 (цианокобаламина) |
| Недостаточность витамина В6 (пиридоксина) |
| Недостаточность витамина Е |
| Недостаточность витамина К |
| Недостаточность никотиновой кислоты (ниацина, витамина РР, витамина В3) |
| Недостаточность селена (дефицит селена) |
| Нейрональный цероид-липофусциноз |
| Непереносимость лактозы |
| Несахарный диабет |
| Ожирение |
| Острый гнойный тироидит (струмит) |
| Острый негнойный тиреоидит |
| Острый тиреоидит |
| Подагра |
| Подострый тиреоидит (тиреоидит де Кервена) |
| Преждевременное половое созревание |
| Псевдогипопаратиреоз |
| Псевдогурлеровская полидистрофия (муколипидоз тип III) |
| Рахит |
| Сахарный диабет 1 типа |
| Сахарный диабет 2 типа |
| Синдром Видемана-Беквита |
| Синдром Грама |
| Синдром Дабина-Джонсона |
| Синдром Деркума |
| Синдром Жильбера |
| Синдром Криглера – Найяра |
| Синдром Лёша-Нихана |
| Синдром Маделунга |
| Синдром монорхизма |
| Синдром поликистозных яичников |
| Синдром Ротора |
| Тиреотоксикоз (гипертиреоз) |
| Тиреотоксический криз (тиреоидный криз) |
| Тирозиноз |
| Фенилкетонурия (фенилпировиноградная олигофрения) |
| Фиброзный тиреоидит (тиреоидит Риделя) |
| Хронический аутоиммунный тиреоидит |
| Энцефалопатия Вернике |